Background
Pathogenic germline variants in SBDS gene are associated with Shwachman-Diamond syndrome (SDS). SDS is classically associated with bone marrow failure and exocrine pancreatic insufficiency and increased risk for developing myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML).1,2 We report three brothers without the classic clinical phenotype of SDS and history of MDS or AML in adulthood.
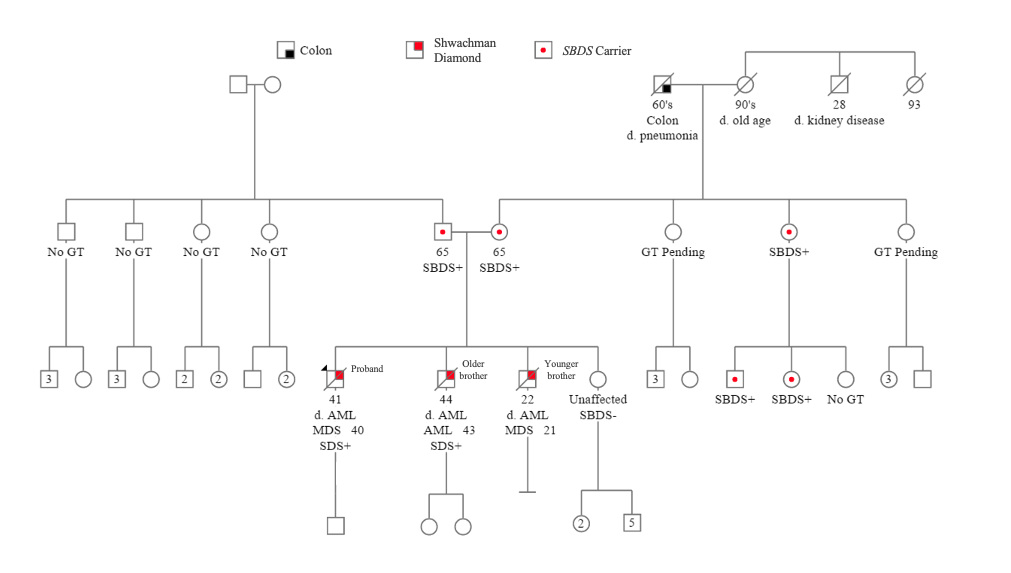
Case discussion
The proband presented with mild persistent neutropenia at age 38. Marrow evaluation showed a markedly hypocellular bone marrow (20%) without a definitive myeloid neoplasm. At age 40, the proband developed worsening cytopenias (Figure 1), and marrow confirmed MDS with excess blasts-2. He was treated with MUD allogeneic stem cell transplant (allo-SCT) and relapsed at day 100. Despite additional treatment, the patient progressed to AML and died at age 41. DNA Sequencing obtained cultured skin fibroblasts identified compound heterozygous pathogenic variants in the SBDS gene.

The proband’s younger brother was diagnosed with MDS at age 21 and underwent allo-SCT. On day 100, he relapsed and had a second allo-SCT. Subsequently, the younger brother progressed to AML and died at age 22. The proband’s older brother presented with pancytopenia at age 43, and marrow evaluation confirmed AML with myelodysplasia-related changes. He was refractory to initial induction chemotherapy and had no response to multiple salvage therapies, and ultimately died at age 44. Site-specific testing of cultured fibroblasts confirmed the same variants in the proband’s older brother. SDS was not confirmed in the proband’s younger brother.
Conclusion
This sibship supports an expansion of the SDS phenotype into adult-onset MDS or AML without classic SDS phenotype. Germline testing for predisposition to myeloid malignancies should be considered in all patients with early-onset MDS/AML, especially in the presence of positive family history or features suggestive of autosomal recessive predisposition syndrome.