Introduction
The therapeutic evolution of non-small cell lung cancer has been dramatic over the past decade with the discovery of oncogenic driver mutations and the advent of biomarker guided management. The journey to the discovery of immune checkpoints dates back to the 1980s, when scientists first discovered T-cell receptors (TCR) which eventually led to the understanding of the major signals required for T-cell activation.1–3 By the mid 90’s, CTLA-4 (cytotoxic T-lymphocyte–associated antigen 4), a homolog of CD28 was discovered which functioned as an inhibitory program on activated T-cells thereby serving as a inhibition to the T-cells.4,5 In 1992, PD-1 (programmed cell death receptor 1), a transmembrane protein similar to CTLA-4, expressed on T-cells was discovered to negatively regulate the immune response of T-cells by interfering with receptor signaling.6 In 1999, B7-H1, also known as PD-L1 (programmed death ligand 1) was discovered and its blockade led to increased levels of IL-10 and IL-2, thereby stimulating T-cells.7 This led to the development of immune checkpoint inhibitors (ICIs). The CTLA-4 inhibitor ipilimumab was approved by the FDA in 2011 for the treatment of untreated metastatic melanoma in combination with chemotherapy after a landmark trial showed an OS benefit.8 The first ICI that was approved for the treatment of lung cancer was nivolumab. CHECKMATE-017 and CHECKMATE-057 were phase 3 trials that were designed to evaluate the efficacy of nivolumab in the management of squamous and non-squamous metastatic NSCLC and both trials showed an OS benefit leading to its approval in previously treated patients.9,10 Similarly in 2016, pembrolizumab was approved after a trial showed an OS benefit in patients with previously treated metastatic NSCLC.11 Currently, various approvals exist for the combination of ICIs with chemotherapy or ICI alone in the front-line management of metastatic NSCLC based on the PD-L1 status.12 The approved immune checkpoint inhibitors include pembrolizumab, atezolizumab, cemiplimab, nivolumab/ipilimumab and durvalumab/tremelimumab. In table 1, we highlight the landmark trials that led to the approval of these agents in the management of metastatic NSCLC.
Despite these advances in the field of lung cancer and immunotherapy, several challenges continue to limit patient outcomes. In this review, we discuss the pressing challenges and controversies in the immunotherapy landscape in non-small cell lung cancer. We first discuss the issues with PD-L1 as an optimal biomarker and segue into the utility of using tumor mutational burden as a biomarker for immune checkpoint inhibitors. We also address other PD1/PD-L1 inhibitors in development and the lack of benefit associated with multiple agents of the same class with similar efficacy and safety profiles. Lastly, we also shine light on newer immunotherapy approaches and inform the reader on the future directions in this field.
PD-L1 expression as a predictive biomarker
There are several mechanisms which regulate the expression of PD-1/PD-L1 proteins in tumor cells (TCs).34,35 PD-L1 is coded by CD274 gene located on chromosome 9p. Mutations in the JAK-STAT pathway result in the amplification and translocation of CD274 leading to its upregulation. Mutations in genes like TP53, KRAS, STK11 and NFE2L2, are known to predict the PD-L1 expression.36 High levels of tumor PD-L1 expression has been shown to corroborate with superior response to ICIs. A positive PD-L1 expression is identified as a TPS ≥1% whereas high PD-L1 expression is identified at a cut off ≥50%. Studies describe a wide range of PD-L1 positivity in NSCLC from 24-60% with prevalence in the <1%, 1-49% and ≥50% groups being 33-60%, 38-42% and 13-28% respectively.37–39
One of the mechanisms of PD-L1 expression is guided by the exposure to IFN-γ released by the T effector cells which increases the expression of PD-L1 on other cellular compartments including tumor infiltrating immune cells (ICs). Therefore, quantification of PD-L1 expression not only includes the tumor proportions score (TPS) but also the degree of PD-L1 expressed on ICs. The significance of immune cells expression of PD-L1 is an evolving subject but it has been hypothesized that the PD-L1 protein of ICs binds with the PD-1 of T cells to facilitate the immune evasion.40
Various FDA approved assays for PD-L1 measurement were compared in the blueprint studies to promote consistency in testing. The assays include: 22C3 pharmDx Dako (pembrolizumab and cemiplimab), SP263 (atezolizumab) and 28-8 pharmDx Dako (nivolumab/ipilimumab). SP142 Ventana assay (atezolizumab) is the only assay approved for testing of PD-L1 expression on ICs.41,42 The Blueprint 1 study demonstrated that the three assays 22C3, 28–8, and SP26 were comparable in their assessment of PD-L1 expression on TCs, whereas the SP-142 PD-L1 assay stained fewer TCs.43 Blueprint 2 confirmed these results and demonstrated that a 5th assay, the 73-10 assay showed greater staining of the TCs. However, when studied in ICs, low level of staining and concordance was noted between all 5 assays.44 In the real world, more cost-effective and easily available laboratory developed tests (LDTs) are also used for assessing PD-L1 expression. The concordance of such tests with the studied assays have not been fully validated and remains a challenge.45,46
Despite the volume of studies correlating response to immunotherapy with PD-L1 expression, this remains a controversial biomarker. Intra-tumoral temporal heterogeneity, spatial heterogeneity and variable concordance between FDA approved assays and LDTs raises questions regarding the predictive and prognostic value of this biomarker.47–50 In KEYNOTE-024, the overall response rate (ORR) of patients with a PD-L1 TPS 50% to pembrolizumab was only 45% meaning that even in patients with a pre-defined high PD-L1 expression, a majority of the population did not respond to the check-point inhibitor as a monotherapy.14 Similarly, in CHECKMATE 227, the objective response rate in the PD-L1 <1% was around 27% and a significant improvement in overall survival was noted with dual check-point inhibition when compared to chemotherapy.18 These varying responses with no clear correlation with the use of dual ICI versus a single ICI adds to the skepticism regarding PD-L1 as a predictive biomarker.
The inter-tumoral discordance in PD-L1 expression of primary tumor when compared metastatic sites particularly the brain has also been highlighted.51–54 Studies have shown that various factors affect tumor heterogeneity such as histology, surgical status, targeted therapy use, prior chemotherapy, immunotherapy use and antibiotic use.55 Herbst et al. demonstrated the dynamic variability of PD-L1 expression in patients treated with atezolizumab, showing an increase in PD-L1 expression with decrease in tumor volume.55,56
The utility of PD-L1 as a predictive biomarker in patients with other driver mutations also has limitations. A meta-analysis showed that the use of ICIs had no OS benefit in the second line setting in NSCLC patients with an EGFR mutation.57 Another phase-2 trial studying pembrolizumab in PD-L1 positive and EGFR mutant NSCLC was ceased due to futility.58 The reason behind the lack of response to ICIs in EGFR mutant NSCLC has been thought to be due to low PD-L1 expression in tumors harboring an EGFR mutation.59 However, there are studies showing conflicting results and others showing a lack of correlation between PD-L1 expression in tumors with driver mutations.60–63
Despite the breadth of data depicting PD-L1 to be an imperfect biomarker, obtaining the PD-L1 expression status is the standard of care in the management of patients with metastatic NSCLC.
Tumor mutational burden as a predictive biomarker for immune checkpoint inhibition
Acquired somatic mutations in tumor DNA can potentially be translated into neo-proteins on the cellular surface which act as neoantigens. These neoantigens are recognized by the T cells using the MHC pathways, subsequently activating the immune cascade. Tumor cells utilize different pathways to evade this immune surveillance, like the PD-1/PD-L1 and CTLA-4 axis. Therefore, in addition to PD-L1, the neoantigen load or the bulk of mutations can also have a predictive role as a biomarker.
Tumor mutational burden (TMB) is defined as the total number of acquired nonsynonymous somatic mutations per megabase of tumor DNA.64 The burden of mutations was initially measured using the whole exon sequencing on both tumor DNA as well as matched normal DNA. Though only non-synonymous mutations lead to changes in protein structures thereby contributing to tumor immunogenicity, synonymous mutations have also been used in the calculations of TMB by different platforms.65 The rationale to include all mutations is to improve the resolution of TMB (using the synonymous mutations as a surrogate for total mutations burden), especially in samples with lowed DNA load.65 Recently specific gene directed next generation sequencing has been validated for TMB measurement and two panels (for tissue TMB) have been approved by the FDA, F1CDx (pembrolizumab in solid tumors with high TMB) by Foundation Medicine Inc. and MSK-IMPACT by the Memorial Sloan Kettering Cancer Center.
The method of calculation of TMB is variable. Different assays have used different methods resulting in poor reproducibility of the results.66 Several assays use proprietary germline variant datasets for filtering germline mutations and some assays use paired tumor and normal tissue to subtract germline alterations and calculate the TMB. Tumor-only sequencing methods have been shown to overestimate TMB compared to the germline-sequenced and subtracted TMB methods. This may particularly impact minority races that have a low representation in most germline variant libraries.67–69 Other issues with TMB calculation are the inclusion of different mutations types (most assays use single nucleotide variants but some also include insertions and deletions, or synonymous mutations), corrections for formalin induced DNA damage (different assays use different methods for compensations for formalin induced deamination), size of the captured coding regions, and the pre-analytical processing which is different for different assays.65,70–73
Blood based assays (like NCC-GP150) to measure TMB using circulating tumor DNA has been tested as surrogate of tissue samples. Though no assay has been approved by the FDA, significant correlation (both technical and clinical) has been demonstrated between the blood TMB (bTMB) and tumor TMB (tTMB) though some studies have shown a low concordance owing to tumor heterogeneity.74,75 bTMB has been demonstrated to be a predictor of clinical response to ICI in NSCLC cases.76,77 Tumor heterogeneity has been graded using several methods and has been demonstrated to influence both tTMb and bTMB.78 Theoretically bTMB represents a more holistic picture of the cellular and genomic diversity within the tumor tissue, but there are limited studies testing this concept. In a study with 32 operated cases of NSCLC muti-region tTMB was found to correlate with both single region tTMB as well as bTMB and high intra-tumoral heterogeneity cases were found to have higher chances of high-TMB when evaluated using muti-regions of tumor tissue.79
The prognostic role of TMB in NSCLC has also been analyzed in systematic review including eight cohorts. Using different cut offs, amongst the high TMB groups (≥10 mut/Mb or ≥ 243 somatic mutations or ≥20 mut/Mb), ICIs were superior to chemotherapy in terms of ORR, PFS and OS. This finding was not seen in the low TMB subgroup.17,18,29,80 In addition, though PFS has been shown to be higher in subjects with high-TMB in tumor tissue, the impact on OS has not seen similar improvements, thereby questioning the role of tTMB in patient selection.18,81
No concordance between PD-L1 levels (assessed using 22C3 platforms) and TMB (assessed using whole exome sequencing) in NSCLC cases have been found.29,82 Even though there have been no head-to-head comparisons, PFS and ORR amongst patients with high PD-L1 expression has been found to be higher than in patients with high TMB.76,83
Current state of immune-checkpoint inhibitors and the utility of “me too” drugs
While the large number of FDA approved agents available for targeting the PD-1/PD-L1/CTLA-4 pathway has opened the doors in-terms of providing various options for the care of patients, they have also been a source of confusion for many oncologists practicing in the United States with regard to teasing out the differences in these regimens, their approved indications, and maneuvering the nuances in choosing the right ICIs for their patients.84 While there are no head-to-head trials comparing various ICIs, a recent meta-analysis has compared various approved ICI regimens and suggested that there may be differences in outcomes based on the PD-L1 status and metastatic sites of patients.85
There is a palpable need to move the needle forward in the treatment of metastatic NSCLC by unlocking the potential of other immune checkpoints. There have been several ongoing trials targeting other pathways with emerging results. One such pathway that is being studied is the T cell immunoglobulin and ITIM domain (TIGIT). The TIGIT protein expression is mainly in T-lymphocytes and NK cells. It competes with CD226 to deliver an inhibitory signal, thereby causing an immunosuppressive effect by decreasing T-cell activation, function and inhibiton of NK cell activity.86–88 Interim results from the phase-3 trial SKYSCRAPER (NCT04294810) assessing the combination of atezolizumab combined with tiragolumab (anti-TIGIT) in metastatic NSCLC did not meet the co-primary endpoint of PFS. Due to immature OS data, the study is being continued. The recently presented interim analysis results of the phase-2 ARC-7 study showed a superior PFS and OS of the doublet domvanalimab (anti-PD-1) plus zimberelimab (anti-TIGIT) and the triplet domvanalimab plus zimberelimab plus etrumadenant (A2a/b adenosine receptor antagonist) when compared to domvanalimab alone in patients with PD-L1 high metastatic NSCLC.89 In the phase-2 CITYSCAPE trial, an improvement in PFS was seen with the incorporation of anti-TIGIT tiragolumab plus atezolizumab compared to ICI alone.90
The challenges associated with ICIs in low and middle-income countries (LMIC) are different. The easy availability of ICIs that exist in upper- middle income and high-income countries have not percolated into LMICs due to multiple barriers such as cost, availability, physician preference and lack of ethnic-diverse representation in clinical trials. Ravikrishna et al., recently showed that in a 5-year time period including more than 15,000 patients in India whom were eligible for ICIs, only about 2.8% received them.91 Nazha et al., have described the involvement of Asian representation in trials to be around 6%.92 In China, there are five PD/PD-L1 agents (camrelizumab, sintilimab, tislelizumab, toripalimab, sugemalimab) have been granted approval. However, these drugs have not made their way to many other Asian countries. The Chinese anti-PD-1 drug sintilimab, was recently denied approval by the FDA approval based on the ORIENT-11 trial,93 partially due to a majority of the patients enrolled being non-US based.84 The manufacturer of this drug had proposed a reduction of the prices of other similar drugs in the market by 40-90%, speaking to the much-discussed speculation regarding improvement in competitive pricing with the development of “me too” drugs. However, whether this theory holds true or not is still debatable with existing evidence suggesting that “brand-brand” competition is historically not known to lower the prices of drugs of the same class.94 The emergence of these “me too” drugs while, unlikely to be beneficial in markets such as the United States and European Union, could solve a pressing issue in other countries where ICIs are not easily accessible. Manufacturing companies can seek to include ethnically diverse populations from countries like India, for example, where patient recruitment is unlikely to be a major issue. This will solve the issues of finding a niche for these “me too” drugs while also improving access to equitable care across other countries.
Emerging checkpoint pathways
There continues to be progress in the field of checkpoint inhibition as numerous other immune checkpoints have been identified as potential targets for inhibition and drug development. Table 2 highlights many of the emerging checkpoints.
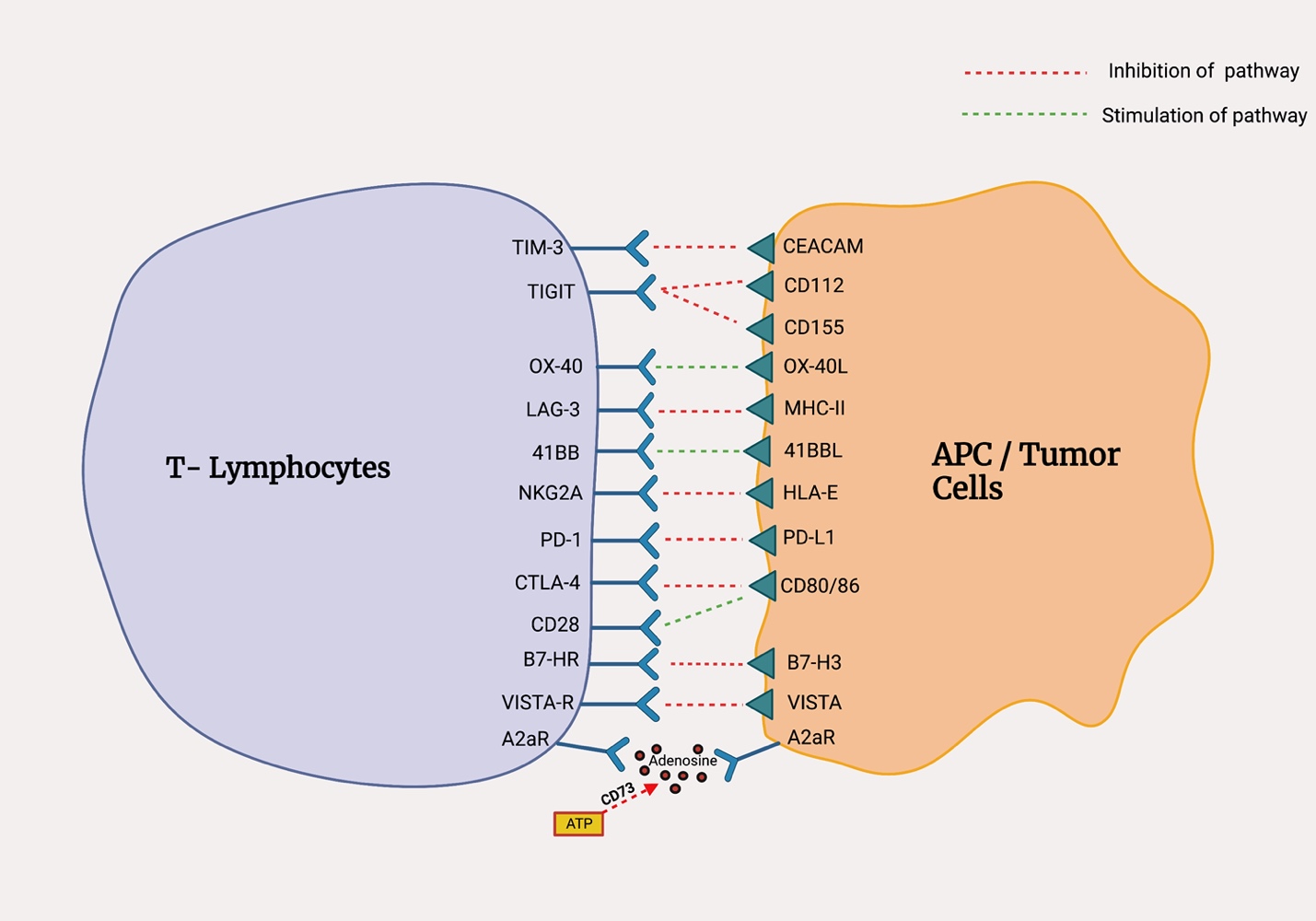
The TACTI-002 (NCT03625323) study studied the combination of the soluble anti-LAG-3 protein, eftilagimod alpha with pembrolizumab in the front-line management of metastatic non-small cell lung cancer showed an ORR in the intention to treat (ITT) population of 37% with a disease control rate was around 73% with more mature data pending.115 Modest results have also been seen in trials studying the combination of TIM-3 with PD-L1 inhibition due to potential synergistic effects of the two drugs. One phase II trial (NCT02608268) studied MBG453 (anti-TIM-3) plus spartalizumab (anti-PD-1) in patients with progressive metastatic NSCLC and demonstrated a durable clinical benefit in about 42% of the NSCLC.116 There are several studies evaluating the anti-tumor effects of 4-1BB agonists alone and in combination with ICIs.12 Two drugs, urelumab (BMS-663513) and utomilumab (PF-05082566) with initial results showed only a modest response in solid tumors.117,118 Monalizumab, an anti-NKG2A humanized monoclonal antibody is being studied in the non-metastatic setting in the management of NSCLC. The interim analysis from the phase-2 COAST study demonstrated promising results with a superior ORR of monalizumab plus durvalumab when compared to durvalumab alone.119 Figure 1 shows newer checkpoints and their ligands.

The road thus far, has been challenging with many of the mentioned therapies showing suboptimal activity when used alone. The synergistic activity of these drugs when used with existing checkpoint inhibitors remains to be further explored with multiple concerns regarding tolerability and more severe immune related adverse events. There is a need for further research in identifying biomarkers and for the development of strategies that can help turn “cold” tumor microenvironments (TME) to “hot” TME, thereby increasing the response to ICIs.121
Conclusion
Immunotherapy in lung cancer still has a long way to go. The immune checkpoint inhibitors, especially PD-1/PDL1 targeted agents, have been shown to significantly improve clinically relevant endpoints. But despite the overwhelming success, patient selection remains an imperfect facet. Research efforts need to be focused on rational combination strategies developed on the basis of improved tumor biology understanding. Different biomarkers with different assays have been tested and approved but the search of an ideal biomarker remains ongoing. Deeper insight into the immune evasion mechanisms of tumor cells might add to the understanding and development of molecules leading to superior outcomes.
Future Directions
In addition to the immune checkpoint inhibitors, various other forms of immunotherapies have also been investigated in different phases of clinical trials like tumor directed monoclonal antibodies, tumor vaccines, T cell therapies, nanomaterials and bi-specific T-cell engagers (BiTEs) and drugs targeting other checkpoints.
Chimeric antigen receptor (CAR)-T cell therapy, part of adoptive cell transfer therapy, utilizes the genetically modified T cells for their actions against tumor cells. Gene coding the modified chimeric proteins are inserted into the autologous T cell using viral or non-viral vectors. These proteins are able to bind to various cell surface antigens or receptors present on tumor cells.122 CAR-T cells have been demonstrated to be successful in hematological malignancies but their response rates in solid tumors have been low, probably due to low levels of tumor infiltrations, lack of tumor specific antigens and risk of cytokine release syndrome.123 Clinical trials for CAR-T cells in lung cancer have tested their role in malignant pleural mesothelioma due to the availability of specific MSLN antigen but role of CAR-T cells in NSCLC has yet not been tested in clinical trials.122 Additionally, TCR (T cell receptor) gene engineered T cells, which have TCR specifically directed towards cancer antigens have also been tested in preclinical models. One of the antigens used for the development of TCRs has been Kita-Kyushu Lung Cancer Antgen-1 which is one of its family proteins, not expressed on healthy tissues.124 PD-1 gene disrupted T cells (using CRISPR-Cas9 technology) have also been tested in phase I trial and demonstrated to be safe and feasible.125 Another form of cellular immunotherapy is TILs (tumor infiltrating lymphocytes). These lymphocytes are usually very few hence, for using this method TILs are expanded in vitro and later reinjected in large amounts. Autologous TIL therapy requires prior lymphocyte depletion. The acceptable safety profile of TIL therapy has been demonstrated in phase 1 study where TILs were administered with nivolumab.126
Bispecific T cell engager (BiTE) platforms is another of targeted immunotherapy where two different antigens are linked together. One end of the protein binds with the tumor antigens whereas the other end binds with the T cells and leads to immune activation.127 AMG 757 (Tarlatamab) is a bispecific T-cell engager targeting delta like ligand 3 (DLCC3) in SCLC. Phase I results of AMG 757 have demonstrated acceptable safety profile and trial is still ongoing.128 Bispecific antibody, Y111, which targets PD-L1 and CD3 has also been tested in preclinical setting where it has been demonstrated to be effective in inducing tumor cell cytotoxicity.129
In addition to the BiTEs, the bispecific monoclonal antibodies which have two different targets have also been discovered and tested. Amivantamab, targeting EGFR and MET has been given an accelerated approval by the FDA for exon 20 insertion mutations, which are inherently resistant to the conventional EGFR TKIs,130 following the results of CHRYSALIS trial which demonstrated an ORR of 40% and DOR of 11.1 months.131 Another example is Zenocutuzumab (MCLA-128) which is a NRG1 fusion targeting bispecific antibody being tested in NRG1 positive solid tumors.132
Apart from the conventional biomarkers, neoantigen load, ctDNA and MSI-H/MMR (mismatch repair) are also being studied as potential biomarkers for ICIs therapy.133 Newer methods of resistance are also being studied and targeted like STK11/LKB1134 and JAK2/STAT.135
Continuing Education Credit Information
The Binaytara Foundation is accredited by the Washington State Medical Association to provide continuing medical education for physicians.
The Binaytara Foundation designates this live activity for a maximum of 1.0 AMA PRA Category 1 Credit™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
This activity meets the criteria for up to 1.0 hours of Category I CME credit to satisfy the relicensure requirements of the Washington State Medical Quality Assurance Commission.
Please click here to register and claim your CME credits.
Conflict of Interest
None
Funding information
N/A
Ethical statements
N/A
Acknowledgement
None
Author contributions
i. All authors: conception and design
ii. All authors: data collection and assembly
iii. All authors: data analysis, manuscript writing
All authors have approved the manuscript.